Lysosomale Speicher-krankheiten
Morbus Hunter, Morbus Fabry und Morbus Gaucher sind sehr seltene genetische Stoffwechselerkrankungen, die immer auf einen genetischen Defekt zurückzuführen sind. Sie werden auch als lysosomale Speicherkrankheiten bezeichnet. Die auftretenden Krankheitsmerkmale sind unspezifisch und nicht bei allen Patienten gleich. Daher werden diese zunächst oft anderen Erkrankungen und nicht den lysosomalen Speicherkrankheiten zugeordnet.
Machen Sie sich ein Bild der Krankheitsmerkmale
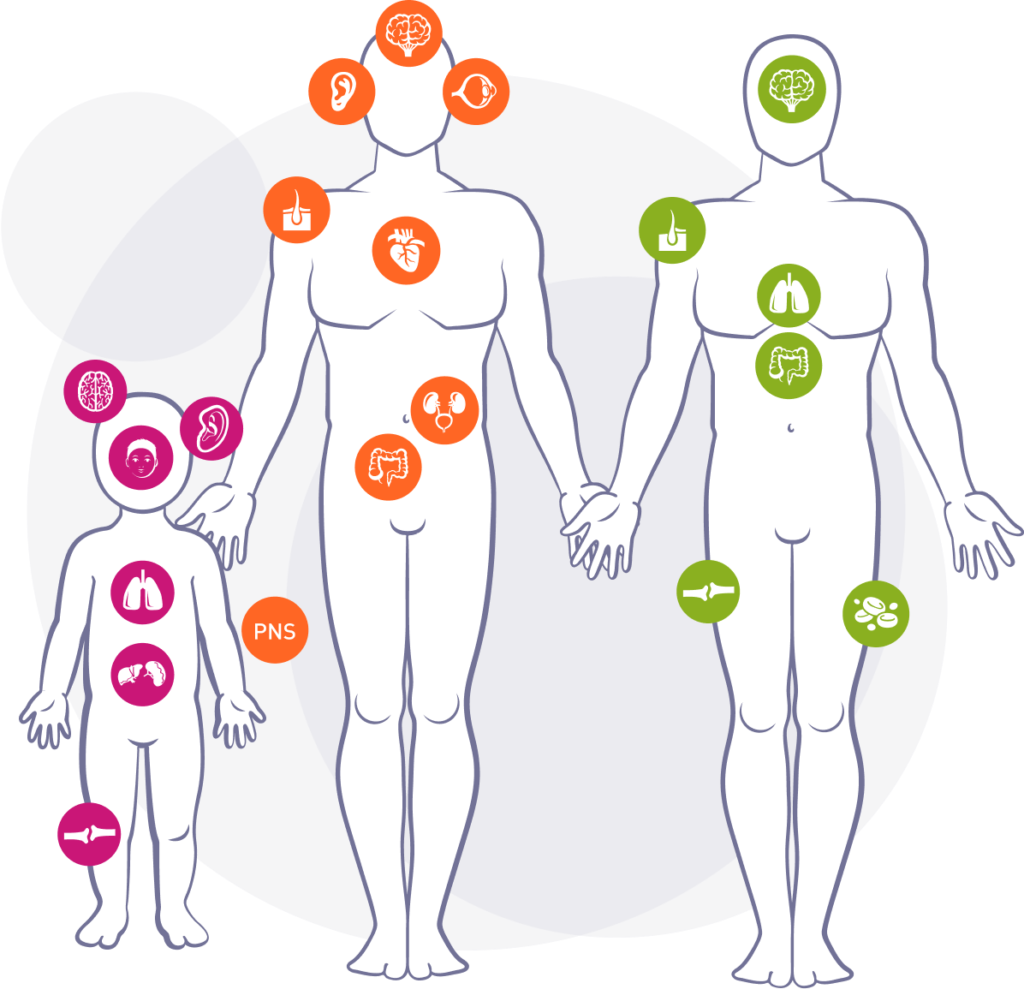
Die auftretenden Krankheitsmerkmale sind bei Morbus Hunter, Fabry und Gaucher sehr unterschiedlich, unspezifisch und nicht bei allen Patienten gleich. Daher ist es sehr schwer, die lysosomalen Speicherkrankheiten auf Anhieb zu erkennen. Auftretende Krankheitsmerkmale werden zunächst oft anderen Erkrankungen zugeordnet.
Morbus Hunter
Betrifft 1 von 162.000 Menschen und ist unter dem Begriff MPS II (auch Mukopolysaccharidose Typ II) bekannt. Betroffen sind von dieser lysosomalen Speicherkrankheit hauptsächlich Buben; ganz selten Mädchen.
Morbus Fabry
Betrifft 1 von 40.000 Menschen. Bei vielen Patienten wird diese Krankheit, die durch eine ungenügende Aktivität eines Enzyms verursacht wird, erst im Erwachsenen-Alter diagnostiziert. Typisch sind brennende Schmerzen an Händen und Füßen.
Morbus Gaucher
Betrifft 1 von 50.000 Menschen. Die häufigsten Krankheitsmerkmale dieser seltenen lysosomalen Speicherkrankheit sind Blutbildveränderungen, Knochenschmerzen und eine vergrößerte Leber und Milz.
Hinweis zum Corona-Virus
Es ist wichtig, dass Sie auf Ihren Körper hören und auftretende Krankheitszeichen, die nicht von selber verschwinden oder sich sogar verstärken, ernst nehmen. Vereinbaren Sie trotz COVID-19 einen Termin bei Ihrem Hausarzt oder Spezialisten, um die Symptome abzuklären! Oft ist auch eine telefonische Kontaktaufnahme möglich, um persönliche Kontakte zu vermeiden.
Falls Sie bereits eine bestätigte Diagnose haben, sollten laufende Therapien und alle Kontrolluntersuchungen trotz COVID-19 fortgeführt werden! Kontaktieren Sie dazu Ihren behandelnden Arzt.
Mehr Informationen und Anlaufstellen in den Bundesländern finden Sie auch beim Öffentlichen Gesundheitsportal Österreich.

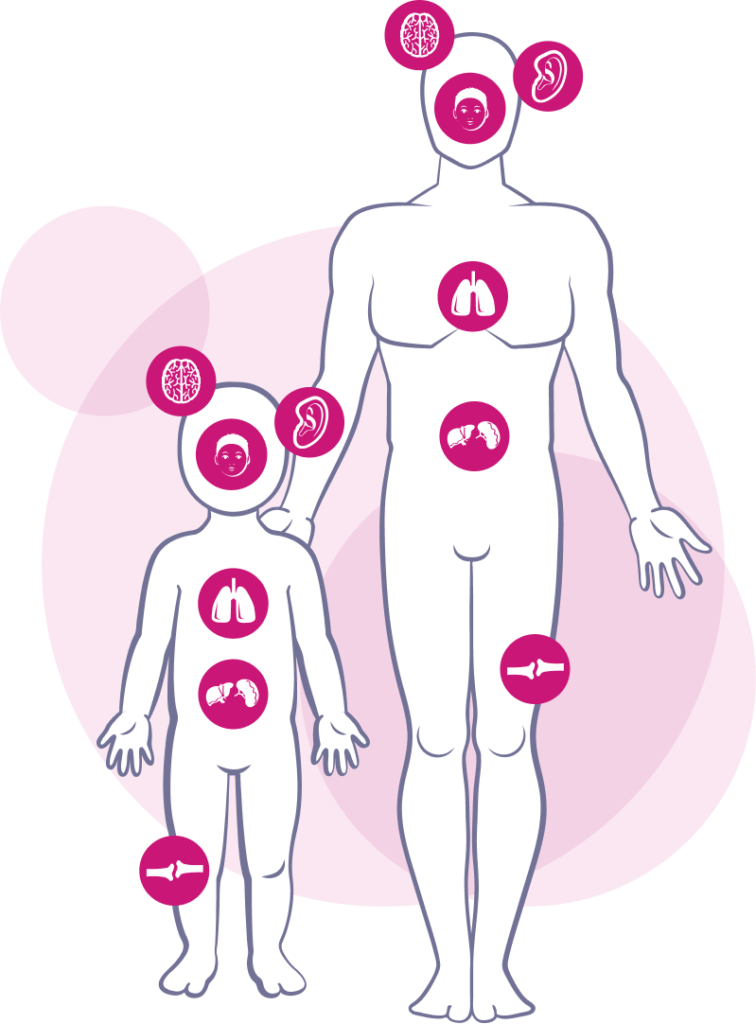
Morbus Hunter
Typisch für diese lysosomale Speichererkrankung, die auch unter MPS Typ II bekannt ist, sind eine ungewöhnliche Kombination normaler Kinderbeschwerden, wie zum Beispiel Leisten- und Nabelbrüche, häufige Mittelohrentzündungen und vergrößerte Mandeln. Charakteristisch sind zudem eine überdurchschnittliche Größe des Kopfes, grobe Gesichtszüge, wiederkehrende Infekte der Atemwege, sowie Krankheiten der Lunge.
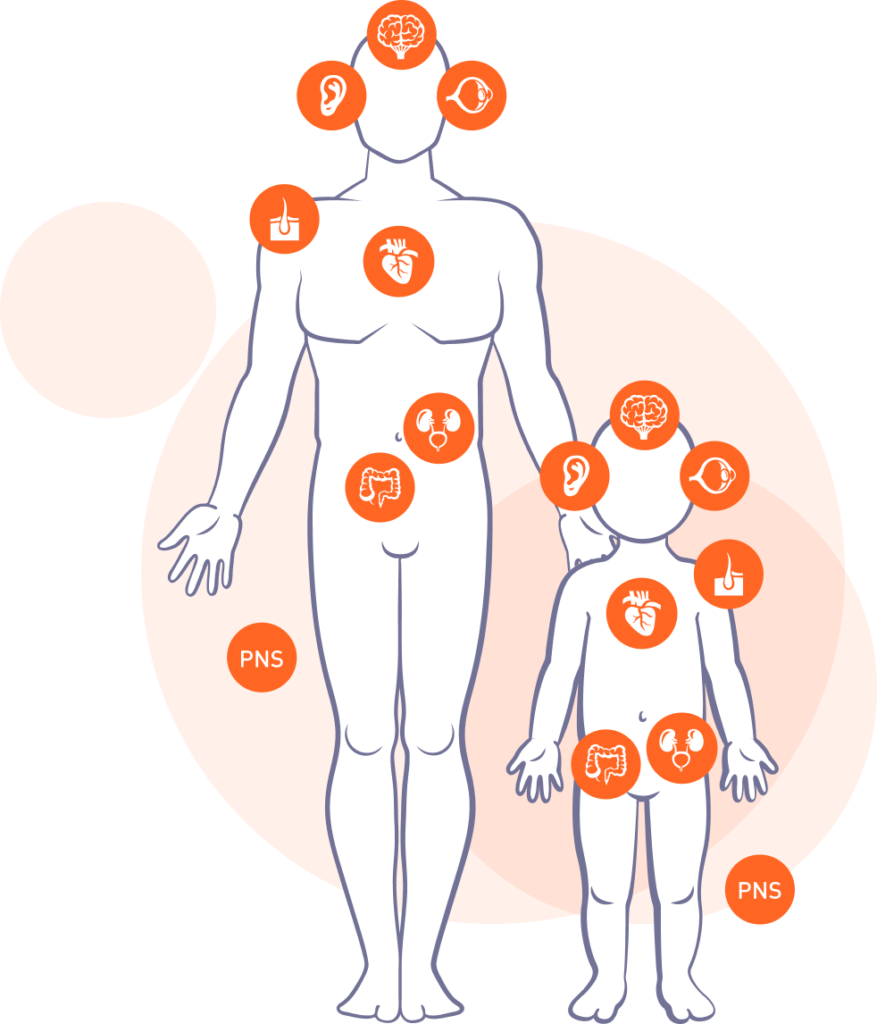
Morbus Fabry
Betroffen sind Nieren, Herz, und Nervensystem sowie Haut, Augen, Gehör und Magen-Darm-Trakt. Charakteristisch bei den Patienten dieser lysosomalen Speicherkrankheit sind anhaltende, brennende Schmerzen in den Händen und Füßen sowie Kälte- und Hitze-Überempfindlichkeit und die Unfähigkeit zu schwitzen.
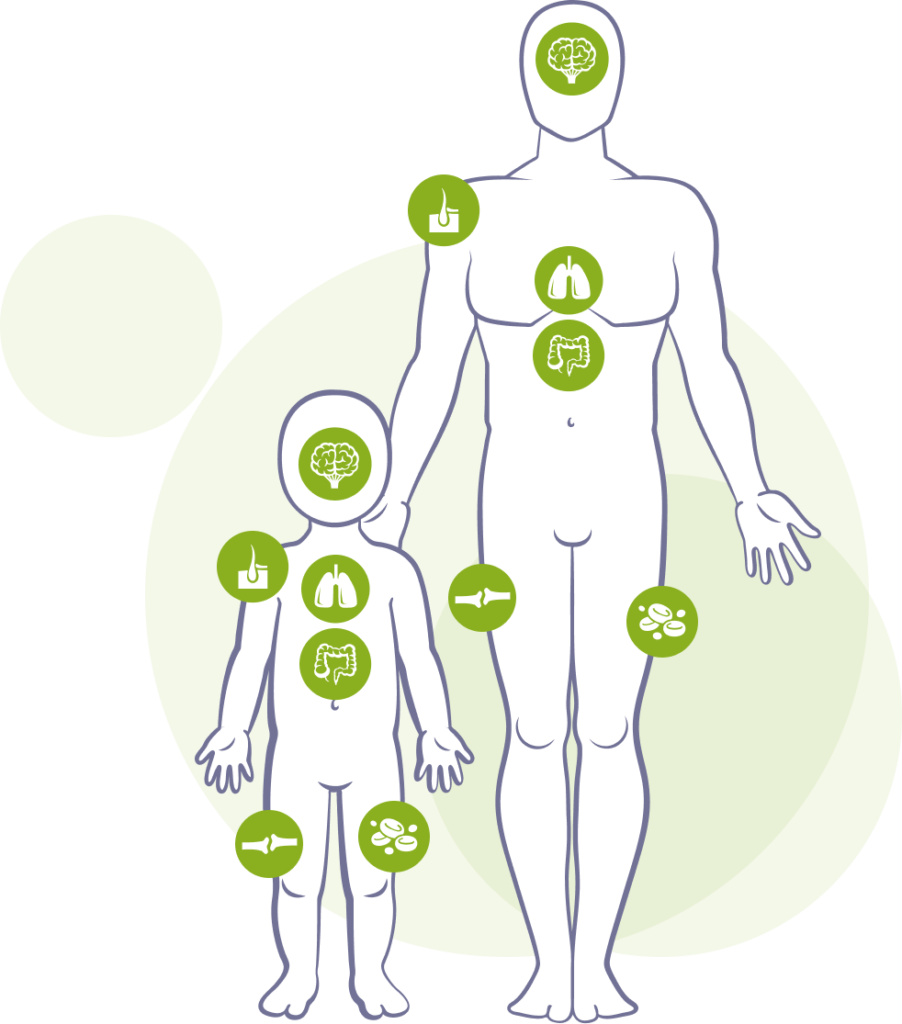
Morbus Gaucher
Typisch sind eine vergrößerte Leber und/oder Milz, Nasen- und Zahnfleischbluten, rasche Entstehung blauer Flecken, Müdigkeit und Knochenschmerzen. Bei Kindern kann es bei dieser Krankheit zu Wachstumsverzögerungen kommen.
Diese Website wurde von Takeda produziert und finanziert und steht der Öffentlichkeit nur zu Informationszwecken zur Verfügung; sie sollte nicht zur Diagnose oder Behandlung von Gesundheitsproblemen oder Krankheiten verwendet werden. Sie ist nicht als Ersatz für die Beratung durch einen Gesundheitsdienstleister gedacht. Bitte kontaktieren Sie Ihren medizinischen Betreuer für weitere Ratschläge. Die Auswirkungen der auf dieser Website beschriebenen Symptome basieren auf den Erfahrungen und der Perspektive einzelner Personen, die mit der Erkrankung leben und sie mit ihren eigenen Worten beschreiben. Nicht alle Menschen, die mit der Erkrankung leben, werden die gleichen Symptome aufweisen.
C-ANPROM/AT/FAB/0006; 01/2021